Todo sobre la fenilcetonuria
La fenilcetonuria (PKU) es una afección poco común en la que un bebé nace sin la capacidad de descomponer adecuadamente un aminoácido llamado fenilalanina.
Causas
La PKU se hereda, lo que significa que se transmite de padres a hijos. Si ambos padres portan una copia no funcional del gen relacionado con esta afección, cada uno de sus hijos tiene un 25 % (1 en 4) de posibilidades de desarrollar la enfermedad.
A los bebés con PKU les falta una enzima llamada fenilalanina hidroxilasa. Es necesario para descomponer el aminoácido esencial fenilalanina. La fenilalanina se encuentra en alimentos que contienen proteínas.
Sin la enzima, los niveles de fenilalanina se acumulan en el cuerpo. Esta acumulación puede dañar el sistema nervioso central y causar daño cerebral.
Síntomas
La fenilalanina desempeña un papel en la producción de melanina en el cuerpo. El pigmento es responsable del color de la piel y el cabello. Por lo tanto, los bebés con esta afección suelen tener la piel, el cabello y los ojos más claros que los hermanos o hermanas que no padecen la enfermedad.
Otros síntomas pueden incluir:
- Retraso en habilidades mentales y sociales.
- Tamaño de cabeza mucho más pequeño de lo normal.
- Hiperactividad
- Movimientos bruscos de brazos o piernas.
- Discapacidad mental
- Convulsiones
- Erupciones en la piel
- Temblores
Si la PKU no se trata, o si se ingieren alimentos que contienen fenilalanina, el aliento, la piel, el cerumen y la orina pueden tener un olor a "ratón" o "a humedad". Este olor se debe a una acumulación de sustancias fenilalanina en el cuerpo.
Exámenes y pruebas
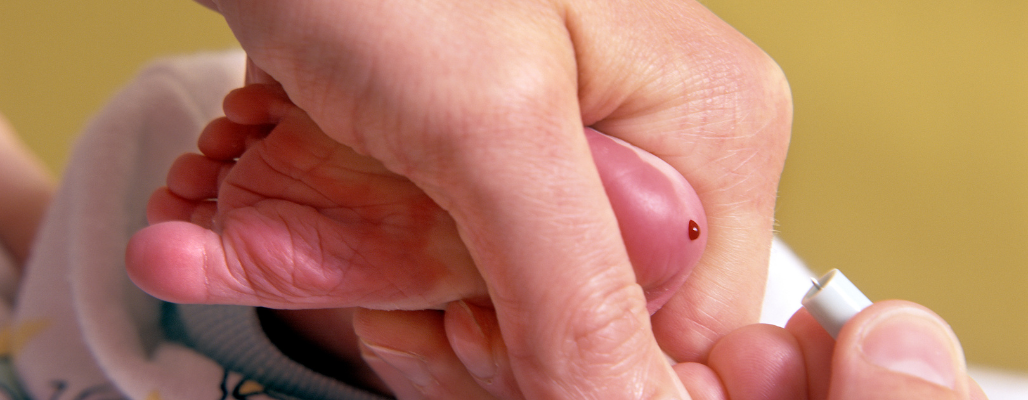
La PKU se puede detectar fácilmente con un simple análisis de sangre. La prueba generalmente se realiza tomando unas gotas de sangre del bebé antes de que abandone el hospital.
Si la prueba de detección es positiva, se requieren más análisis de sangre y orina para confirmar el diagnóstico. También se realizan pruebas genéticas.
Tratamiento
La PKU es una enfermedad tratable. El tratamiento implica una dieta muy baja en fenilalanina, especialmente cuando el niño está creciendo. La dieta debe seguirse estrictamente. Esto requiere una estrecha supervisión por parte de un nutriólogo o médico registrado y la cooperación de los padres y el niño. Quienes continúan la dieta hasta la edad adulta tienen mejor salud física y mental que quienes no la siguen. La "dieta de por vida" se ha convertido en el estándar que recomiendan la mayoría de los expertos. Las mujeres que tienen PKU deben seguir la dieta antes de la concepción y durante todo el embarazo.
Hay grandes cantidades de fenilalanina en la leche, los huevos y otros alimentos comunes. El edulcorante artificial NutraSweet (aspartamo) también contiene fenilalanina. Se debe evitar cualquier producto que contenga aspartamo.
Existen varias fórmulas especiales elaboradas para bebés con PKU. Estos se pueden utilizar como fuente de proteínas extremadamente baja en fenilalanina y equilibrada para los aminoácidos esenciales restantes. Los niños mayores y los adultos utilizan una fórmula diferente que proporciona proteínas en las cantidades que necesitan. Las personas con PKU necesitan tomar fórmula todos los días durante toda su vida.
Pronóstico
Se espera que el resultado sea muy bueno si se sigue estrictamente la dieta, empezando poco después del nacimiento del niño. Si el tratamiento se retrasa o la afección no se trata, se producirá daño cerebral. El funcionamiento escolar puede verse ligeramente afectado.
Si no se evitan las proteínas que contienen fenilalanina, la PKU puede provocar discapacidad mental al final del primer año de vida.
Posibles complicaciones
Se produce una discapacidad mental grave si el trastorno no se trata. El TDAH (trastorno por déficit de atención con hiperactividad) parece ser un problema común en quienes no siguen una dieta muy baja en fenilalanina.
Prevención
Un ensayo enzimático o una prueba genética pueden determinar si los padres son portadores del gen de la PKU. Se puede realizar una muestra de vellosidades coriónicas o una amniocentesis durante el embarazo para detectar PKU en el feto.
Es muy importante que las mujeres con PKU sigan estrictamente una dieta estricta baja en fenilalanina tanto antes de quedar embarazadas como durante todo el embarazo. La acumulación de fenilalanina dañará al bebé en desarrollo, incluso si el niño no ha heredado la enfermedad completa.